










中国医療機器登録管理制度は、経済、社会の発展、特に医療機器産業の発展と共に、改善しつつある。2000年1月、中国国務院は「医療機器監督管理条例」(国務院令第276号)を公布した。同年4月1日より、当該法令は実施することを契機に、中国医療機器監督管理は全面的に遵法行政、遵法監督管理の新世代に入り、医療機器登録管理の法規体系と運行体系は徐々に確立され、且つ改善していく。これは、国民の医療機器応用安全と医療機器産業の健康な発展を著しく促進した。
ここで、中国医療機器登録管理の状況を簡単に説明する。
1 医療機器登録管理法規の発展の歩み
普通の生活用品と異なり、医療機器を使用する時の安全性を確保するために、関連法規は医療機器の市場アクセスを判定する時の重要な根拠と決定因子になる。世界最初の全面的な医療機器法規はアメリカ食品医薬品局(FDA)より1976年に公布された連邦食品・医薬品・化粧品法(Federal Food, Drug, and Cosmetic Act、 FDCA)である。数回の改定は実施されたが、この法規は医療機器の製造と設計管理に大きな影響を与えつづけている。 FDCAは医療機器に対する要求事項はファーマアプローチPharmaceutical Approachに基づき、市販された医療機器の管理経験に基づき、製品の具体的な査察ガイドラインを制定した。もう1つの重要な影響のある医療機器法規は1993年に欧州で公布された「医療機器指令」(MDD: Medical Device Directive 93/42/EEC)で、医療機器の安全性と有効性に対する要求がより広く、細かくなった。且つ、付録の中に基本用件(Essential Requirements:ERs)を規定し、医療機器の安全性を判定する重要な根拠になっている。その他に、欧州医療機器指令は「整合規格」(Harmonized Standards)を提唱し、整合規格の中の技術仕様は市販前審査の重要な根拠に位置づけ、医療機器指令に要求された基本要件(ERs)を満たすために、医療機器製造業者は関連規格に従わなければならない。より深い意味あるのは、医療機器法規を医薬品法規の外延に位置づけさせるやり方と異なり、欧州医療機器指令は独立の法規で、医薬品法規と完全に違うエンジニアリングアプローチ(Engineering Approach)を採用していることである。

「製品(Product)」と「用途(Use)」は医療機器の安全性と性能を決定する医療機器の規制の重要な要素である。製品の安全性と性能に関しては、市販前の審査(市場アクセス)でコントロールし、使用後の持続的な安全性と有効性については市販後のモニタリングで確保できる。世界保健機関(WHO)は、医療機器の管理を動態的なプロセスを見なすことを薦めている。そのライフスパンは図1に示している。
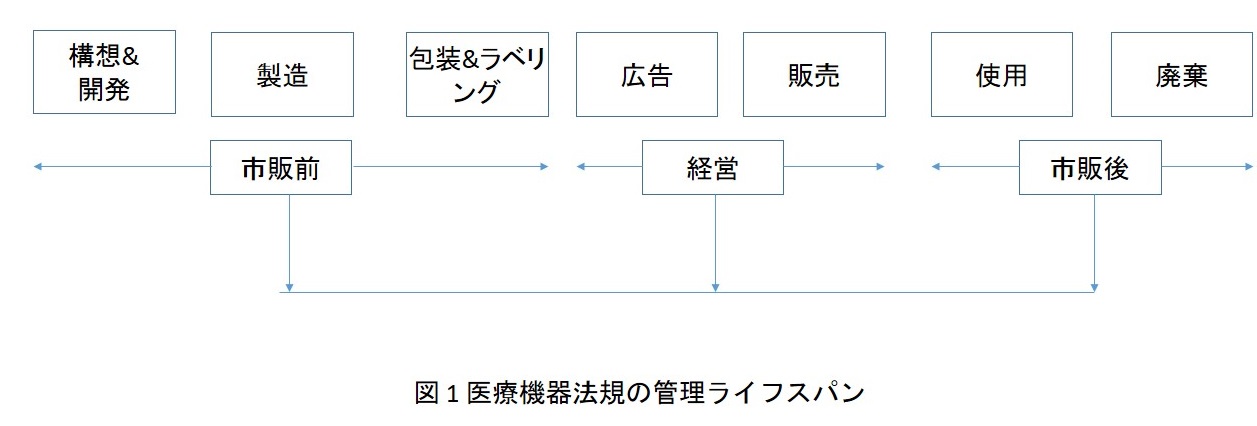
従い、医療機器の市場アクセスは市販前の審査に対する法規の規定による。
4ページ中 1ページ目
執筆者について

李 非
経歴 1999年、清華大学卒業後、ペンシルベニア大学生物医学大学院入学。2003年大学院卒業後、中国瀋陽Philips and Neusoft Medical Systems Co., Ltdに入社、医療機器開発に従事。2005年より遼寧省食品医薬品監督管理局技術審査評価センター医療機器部門長。 ※このプロフィールは掲載記事執筆時点での内容となります








コメント
/
/
/
この記事へのコメントはありません。
コメント